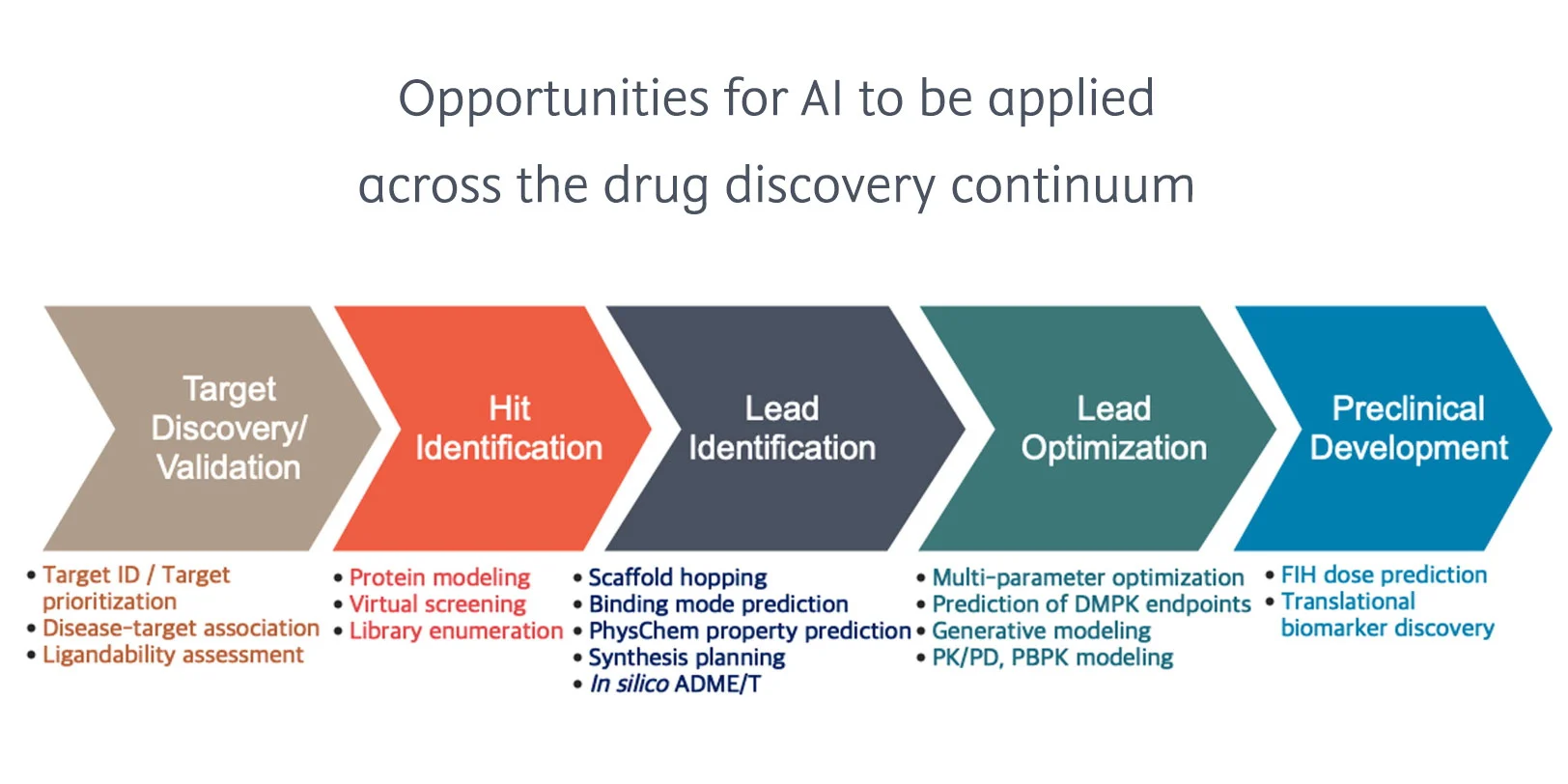
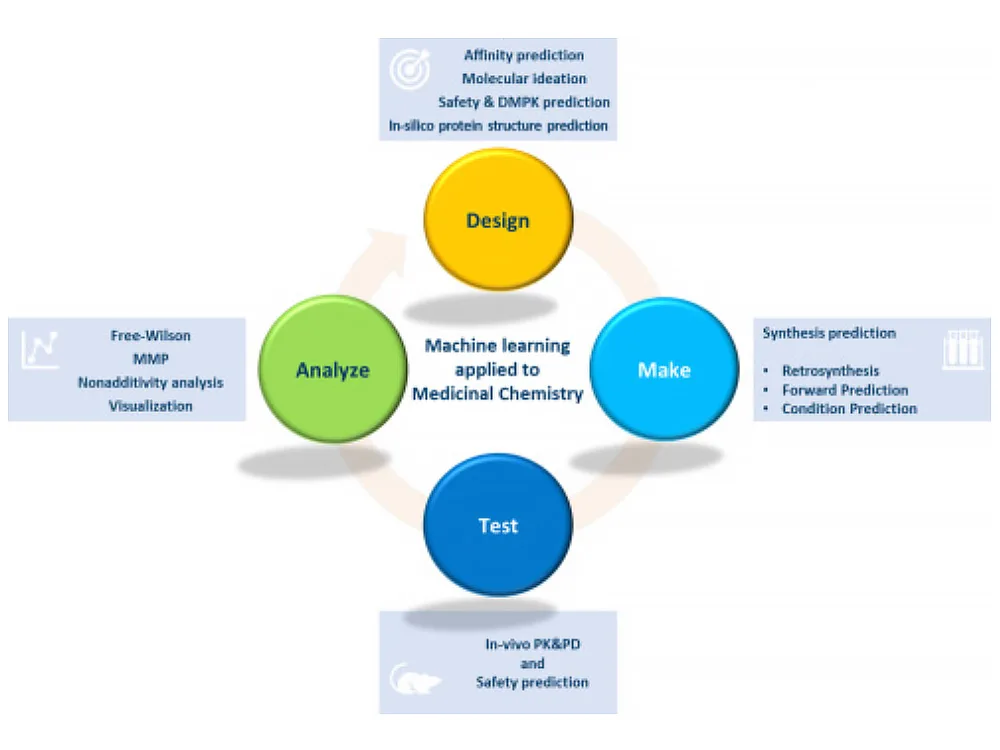
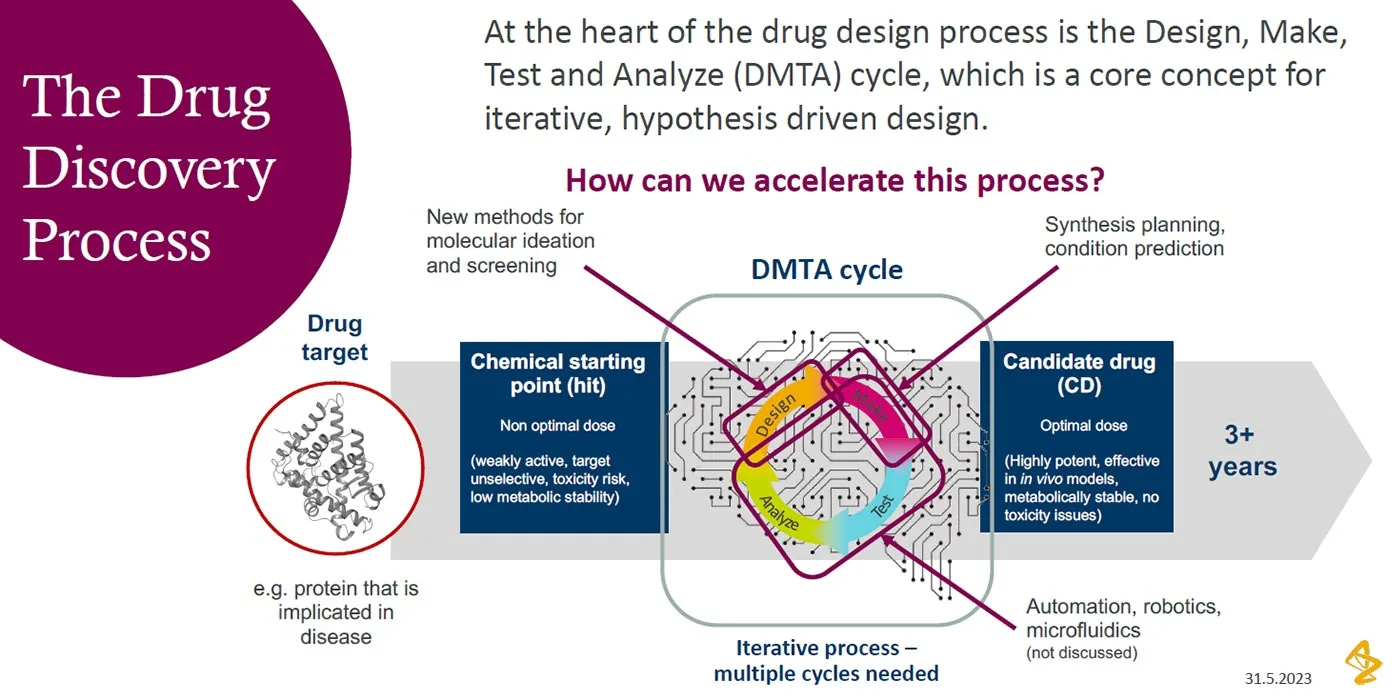
The evolution of AI in small molecule drug discovery
Traditionally, it takes a lot of time and scientific expertise to discover and synthesize a small molecule that becomes a preclinical candidate. But advances in AI are reinventing the processes involved and accelerating drug discovery. Chemists must transform alongside this shift, acquiring the skills and knowledge to apply AI in their work while collaborating more closely than ever with computational chemistry and data science teams.
In a short space of time, artificial intelligence (AI) and associated techniques like machine learning (ML) and generative AI (GenAI) had a considerable impact on chemistry and small molecule discovery. One study1 found that, “…biotech companies using an AI-first approach have more than 150 small molecule drugs in discovery and more than 15 already in clinical trials.”
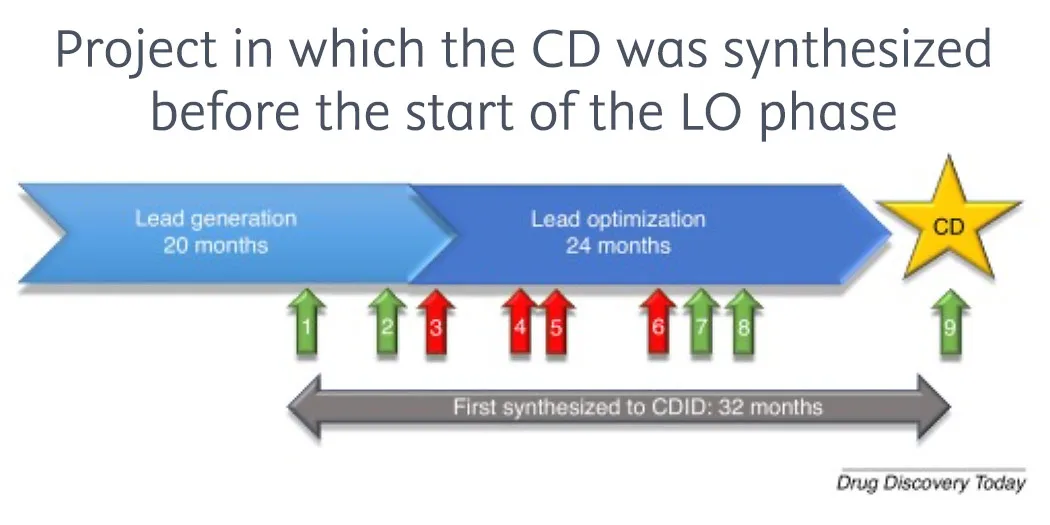
Estimates2 suggest it takes 10–15 years and typically costs up to US $2.8 billion to develop a new drug, and a huge proportion (80–90%) of candidates fail in the clinic. The early discovery stage alone takes an average3 of 3–6 years and accounts for 42% of total capitalized costs in the development of a new drug. Given the cost and time of creating a drug, it’s not surprising that interest in AI-enabled small molecule drug discovery is growing.