Comment réaliser une Échographie pelvienne de l’enfant et de l’adolescente ?
15 décembre 2017
Par Anne-Claire Nonnotte
Nous vous proposons de découvrir un article de l'ouvrage Échographie et imagerie pelvienne en pratique gynécologiqueopens in new tab/window

Échographie et imagerie pelvienne en pratique gynécologique
Échographie pelvienne de l’enfant et de l’adolescente
S. Chapelière, A. Mathiot, S. Franchi-Abella, C. Adamsbaum44. Avec la collaboration pour les éditions précédentes de : A.-E. Millischer, C. André, G. Kalifa.
L’échographie reste la meilleure technique d’imagerie pour l’étude du pelvis de l’enfant, de la naissance à la puberté. Sa réalisation est simple, nécessitant parfois chez le petit enfant une contention, mais jamais de sédation. Une sonde sectorielle de 5 MHz suffit le plus souvent. Chez le nouveau-né, la sonde de 7,5 MHz peut être utilisée et chez l’adolescente, la sonde de 3,5 MHz est souvent utile. L’examen par sonde endovaginale est proscrite chez les jeunes filles vierges. Chez le jeune enfant, la seule difficulté est d’obtenir une réplétion vésicale suffisante pour visualiser le corps utérin et les ovaires qui sont haut situés dans le pelvis. Chez le petit, on peut donner un biberon avant l’examen et éviter une miction trop rapide en « glissant » la sonde sous la couche sans trop le déshabiller. La sensation de froid fait en effet très souvent uriner les nourrissons.
À tout âge, il faut être patient et n’effectuer l’examen que lorsque la vessie est suffisamment pleine. Un repérage initial, par des coupes transversales, de la situation médiane ou latérale de l’utérus permet d’effectuer une coupe sagittale strictement dans l’axe utérin et de mesurer au mieux la hauteur utérine. L’épaisseur du corps utérin est appréciée en coupes sagittales et transversales. C’est cette épaisseur qui se modifie lors du développement utérin alors que celle du col reste quasiment stable. Les ovaires sont repérés en coupes transversales et longitudinales en arrière de la vessie, en dedans de la paroi musculaire du pelvis et en avant des vaisseaux iliaques internes. Leur volume peut être estimé selon la formule approchée : V = 1/2 × longueur × largeur × épaisseur. En pratique, on se contente de la mesure la plus exacte possible du grand axe de l’ovaire. Dans certaines indications, l’échographie peut être complétée par voie périnéale (cf. § Anomalies de la différenciation sexuelle). L’IRM est pratiquée en complément de l’échographie, pour la caractérisation des masses pelviennes, pour l’exploration, chez les jeunes filles, de syndromes des ovaires micropolykystiques et de plus en plus pour les bilans de dysménorrhées sévères à la recherche d’endométriose. Les examens par scanner sont de plus en plus rares et réservés aux bilans d’extension de masses pelviennes ou pour la caractérisation de calcifications au sein des tissus.
Anatomie échographique normale
La connaissance des aspects échographiques des organes génitaux internes chez la fille, de la naissance à la puberté achevée, a permis de définir des critères de développement utéro-ovarien et de différencier ainsi les situations cliniques pathologiques des variantes du développement.
Utérus
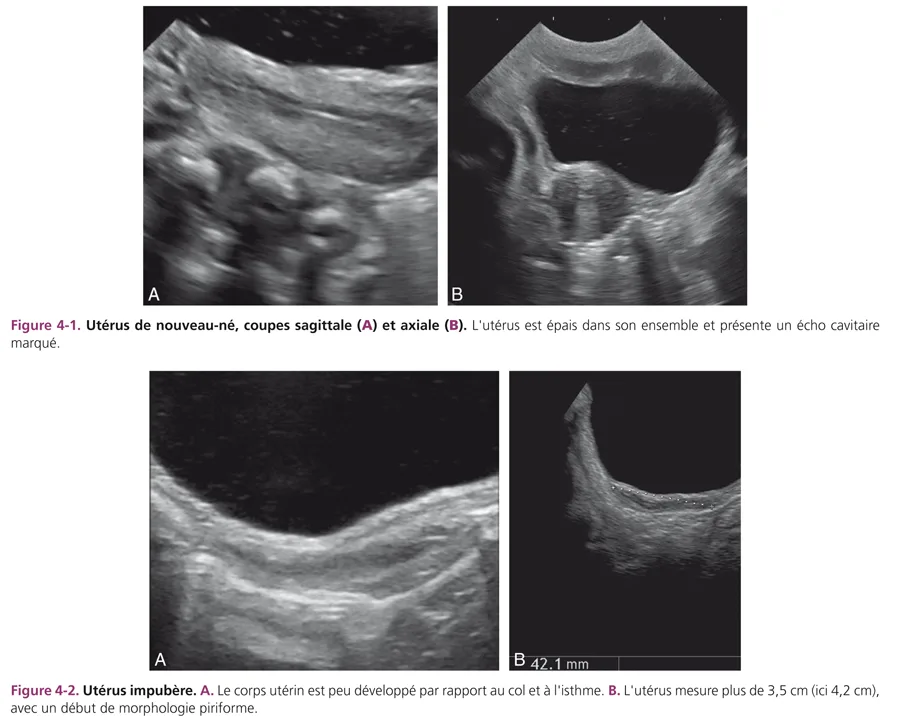
L’utérus des nouveau-nés paraît volumineux du fait de la petite taille du pelvis et de son développement transitoire dû à l’imprégnation normale maternelle pendant la grossesse. Il est épais avec un corps aussi épais que le col, mesuré de 1 à 1,5 cm (fig. 4-1). Sa hauteur est de 3,5 à 4,5 cm. La cavité utérine est soulignée par un écho dense médian, témoin d’un léger développement de l’endomètre. Le vagin sous-jacent présente des parois musculaires hypoéchogènes épaisses, bien visibles. Cet aspect de gros utérus à la période néonatale disparaît en quelques semaines ou mois pour adopter la morphologie impubère normale [1–3]. L’utérus impubère a une morphologie particulière « en goutte » avec un corps utérin fin par rapport au col et à l’isthme plus épais, formant les deux tiers du volume utérin (fig. 4-2A). Le rapport corps sur col est inférieur à 1. La hauteur utérine est mesurée à 3 cm ± 0,5 cm. La taille et l’aspect de l’utérus restent inchangés jusqu’à l’âge de 8 à 9 ans. Son échostructure est hypoéchogène, homogène. La ligne cavitaire n’est pas visible. Un fin écho cavitaire est parfois vu au niveau du col. Les parois vaginales sont fines.
À la puberté (fig. 4-2B), sous l’influence des œstrogènes, l’utérus grossit progressivement et sa croissance s’effectue au niveau du corps qui s’allonge et s’épaissit. L’utérus devient ainsi tubulé puis prend son aspect piriforme pubère. En fin de puberté, il mesure 6 à 6,5 cm de hauteur. La cavité utérine est visible sur toute la hauteur utérine, ce qui témoigne de l’épaississement des parois musculaires, puis de l’épaississement progressif de l’endomètre.
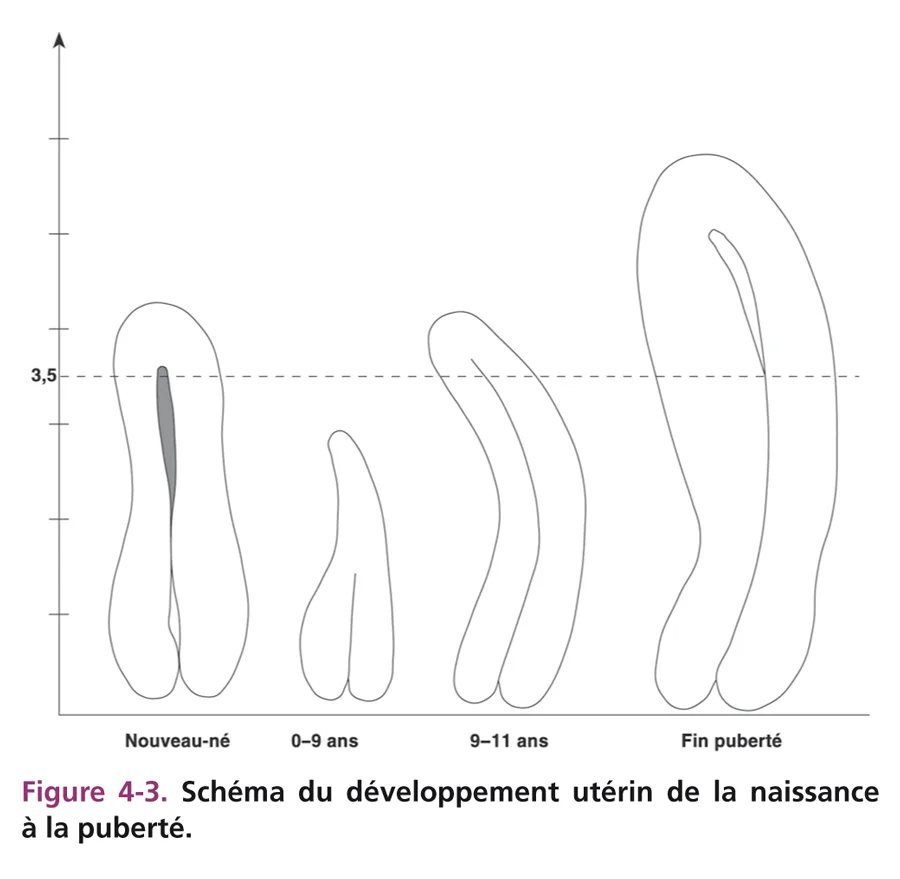
Les critères échographiques du développement utérin par imprégnation hormonale sont donc : ■ hauteur utérine supérieure à 3,5 cm ; ■ épaississement du corps avec un rapport corps/col supérieur à 1 ; ■ visibilité d’une ligne cavitaire échogène sur toute la hauteur utérine.
Ces modifications utérines pendant l’enfance sont schématisées sur la figure 4-3.
Ovaires
Les critères du développement ovarien tenant compte de la taille et de l’échostructure de l’ovaire sont plus difficiles à définir. Rappelons la difficulté à visualiser l’ovaire, souvent haut situé dans le pelvis, nécessitant une bonne réplétion vésicale. Si l’utérus est latérodévié, l’ovaire du même côté est difficilement perçu. Il peut être en position sus- ou rétroutérine (fig. 4-4). À la période néonatale, la taille de l’ovaire est fonction du développement folliculaire lié à la stimulation hormonale gonadotrope maternelle. Plusieurs follicules (taille supérieure à 5 mm) sont visibles, hypo- ou anéchogènes (fig. 4-5). Comme pour l’utérus, cet aspect est transitoire. Les kystes folliculaires (diamètre supérieur à 20 mm) sont fréquents pendant la grossesse et à la période néonatale.

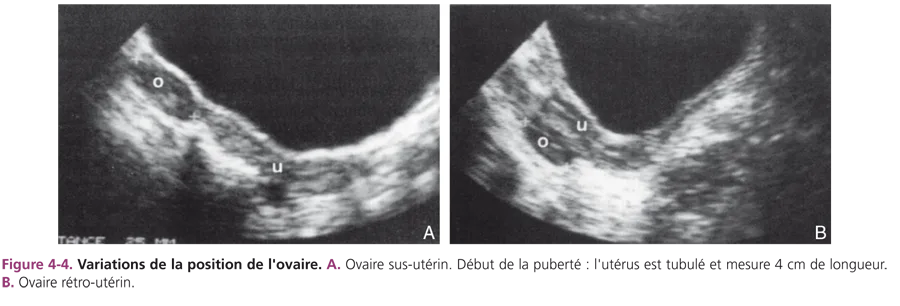
Fig 4-4
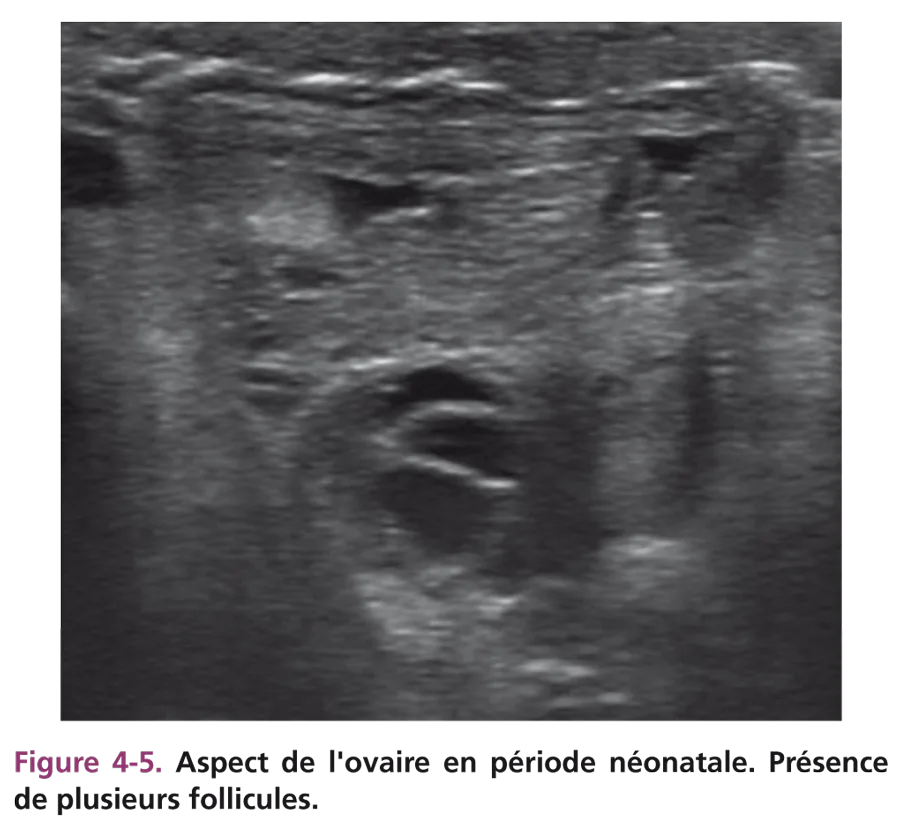
Fig 4-5
Pendant l’enfance, la taille de l’ovaire croît régulièrement. Chez la fillette avant 4 ans, le volume ovarien est en général inférieur ou égal à 1 cm3, soit en mensurations moyennes : 2 cm de longueur × 1 cm de largeur et 1 cm d’épaisseur. Entre 6 et 8 ans, la longueur ovarienne excède souvent 2,5 cm. À partir de 9 ans, l’ovaire mesure 3 cm ou plus. Le développement ovarien précède celui de l’utérus et apparaît antérieur à l’apparition des premiers signes cliniques du développement pubertaire (fig. 4-6). Chez l’enfant impubère, l’ovaire se présente comme une structure ovoïde, hypoéchogène, parfois homogène, mais le plus souvent hétérogène, en rapport avec l’existence de microfollicules dont la taille reste inférieure à 5 mm. Cet aspect microfolliculaire se retrouve chez 93 % des filles entre 2 et 11 ans. À tout âge, il est normal d’observer des follicules ovariens et leur nombre augmente avec l’âge. La constatation d’ovaires plurifolliculaires en période prépubertaire est banale [3–5]. À la puberté, les ovaires sont considérés comme stimulés, développés, lorsque leur longueur est supérieure à 3 cm ou leur volume supérieur à 3 cm3 et qu’ils présentent une activité folliculaire avec visibilité de plusieurs follicules. Cet aspect, associé à un développement utérin, marque la puberté en cours. Après la puberté, pendant les premières années de menstruations, il est habituel de constater un aspect d’ovaires multifolliculaires, avec présence de nombreux follicules de taille variable, occupant le volume ovarien. Cet aspect, très probablement lié aux cycles initiaux anovulatoires (fig. 4-7), est tout à fait normal.
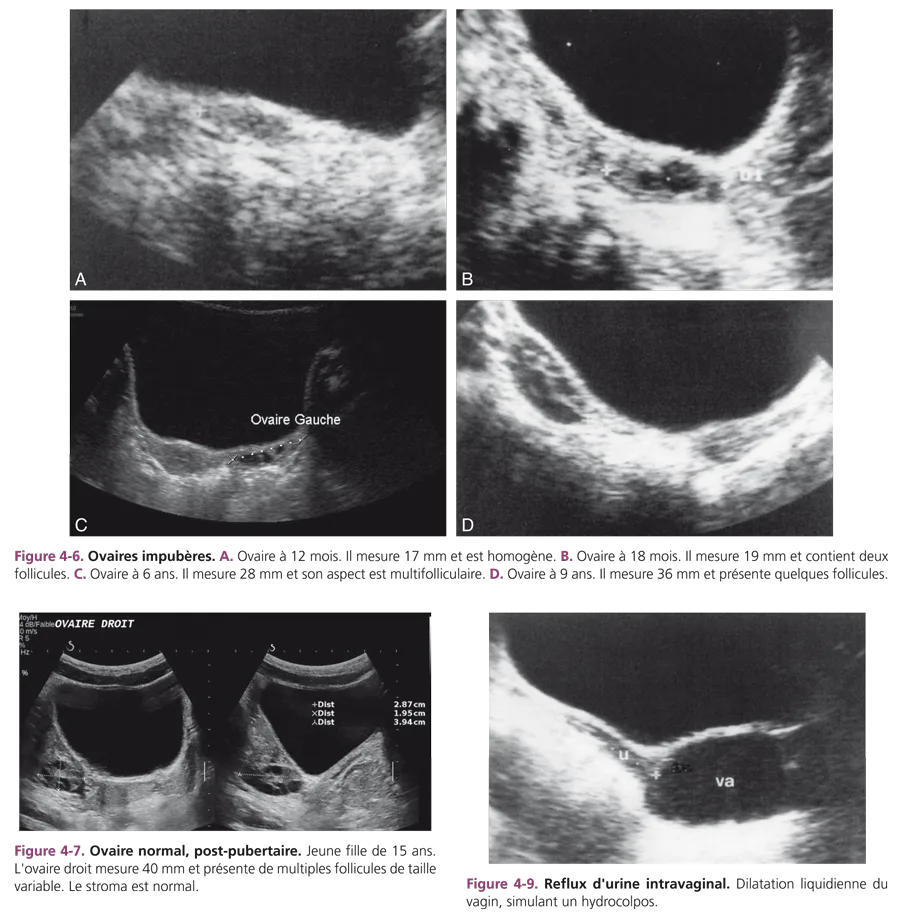
La constatation d’un épanchement liquidien du cul-desac de Douglas est fréquente, surtout chez les petites filles impubères. Il est vraisemblablement lié à l’hydratation orale rapide (fig. 4-8).
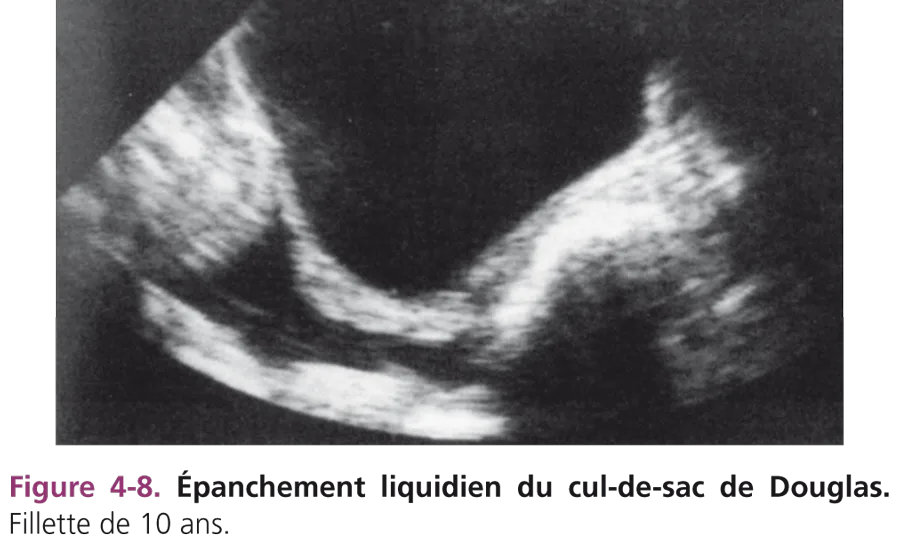
Enfin, l’existence d’un reflux d’urine dans le vagin est banale. Il peut simuler un hydrocolpos mais disparaît après miction ou en position debout (fig. 4-9). Les principales indications de l’échographie pelvienne chez l’enfant sont mentionnées dans l’encadré 4-1.

Anomalies du développement pubertaire
Rappel du développement pubertaire normal
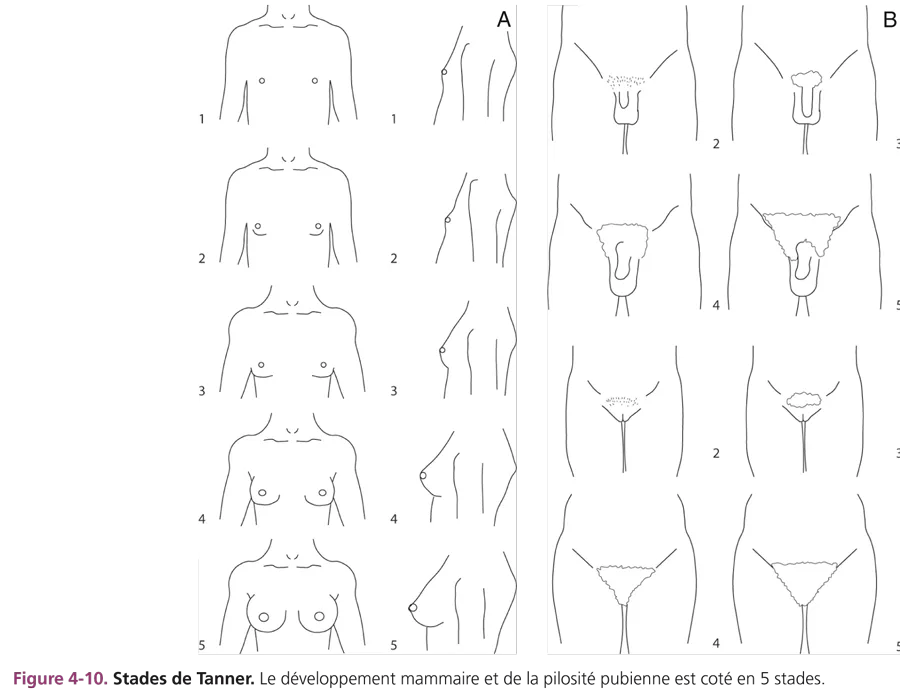
L’apparition des caractères sexuels secondaires survient entre 8,5 et 13 ans chez 95 % des filles et débute par une poussée mammaire. Pour mémoire, chez 95 % des garçons, le début de la puberté dont témoigne le développement testiculaire se situe entre 10 et 13,5 ans. L’accroissement progressif de la pilosité pubienne, le développement mammaire, testiculaire et pénien sont cotés selon les 5 stades de Tanner (fig. 4-10). La durée moyenne entre la poussée mammaire initiale et les menstruations est d’environ 2 ans. Chez le garçon, le développement pubertaire est en moyenne complet en 3 ans. Parallèlement, se produit une accélération de la vitesse de croissance staturale avec une maturation osseuse progressive cotée sur la radiographie de la main selon l’atlas de Greulich et Pyle.
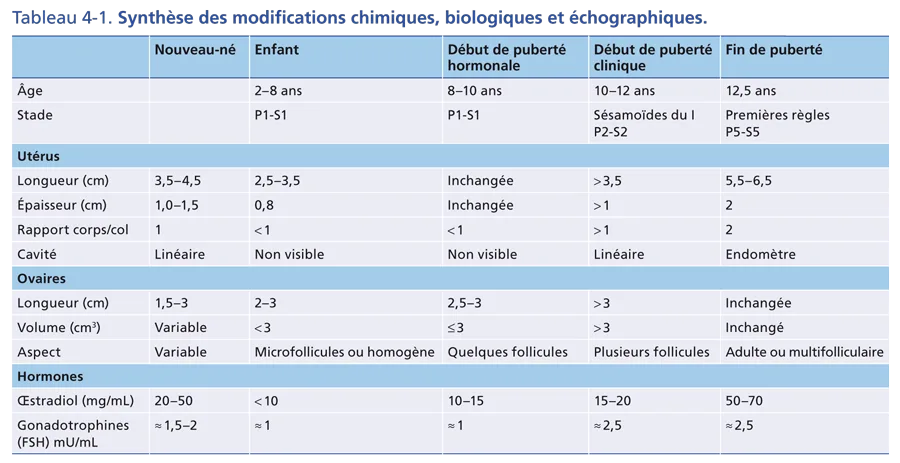
Sur le plan biologique, l’activation de l’axe gonadotrope est marquée par une sécrétion pulsatile de GnRH (Gonadotropin Releasing Hormone) hypothalamique qui provoque une sécrétion également pulsatile de gonadotrophines hypophysaires. L’élévation du taux plasmatique des stéroïdes sexuels, œstradiol et testostérone, est concomitante. L’ensemble des modifications chimiques, biologiques et échographiques est résumé dans le tableau 4-1.
Puberté précoce La précocité pubertaire se définit comme l’apparition des caractères sexuels secondaires avant 8 ans chez la fille et 10 ans chez le garçon. Elle est bien plus fréquente chez la fille que chez le garçon. Dans le bilan pratiqué, la place de l’échographie pelvienne est prépondérante car cet examen simple et reproductible permet de juger rapidement du degré du développement utéro-ovarien et de distinguer ainsi les vrais développements pubertaires des variantes cliniques du développement pubertaire, partielles et régressives, sans signification pathologique.
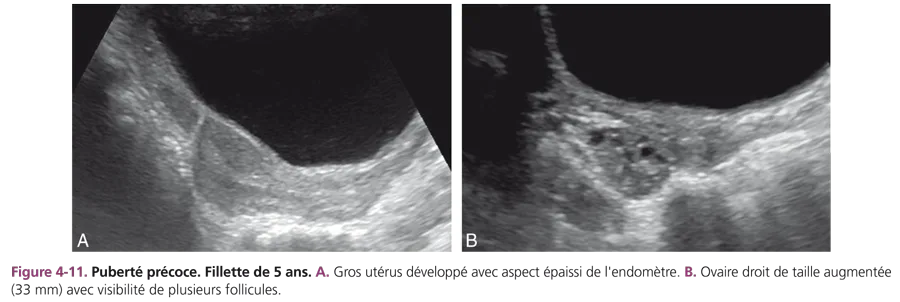
Puberté précoce centrale Elle touche 9 filles pour 1 garçon et est très souvent idiopathique (plus de 90 % des cas). Elle est suspectée cliniquement devant un développement harmonieux des caractères sexuels secondaires, une accélération de la vitesse de croissance et une avance de l’âge osseux. L’échographie pelvienne montre un développement utéro-ovarien plus ou moins évolué (fig. 4-11). Des kystes folliculaires d’hyperstimulation peuvent être visualisés dont l’échographie contrôle le caractère rapidement régressif.
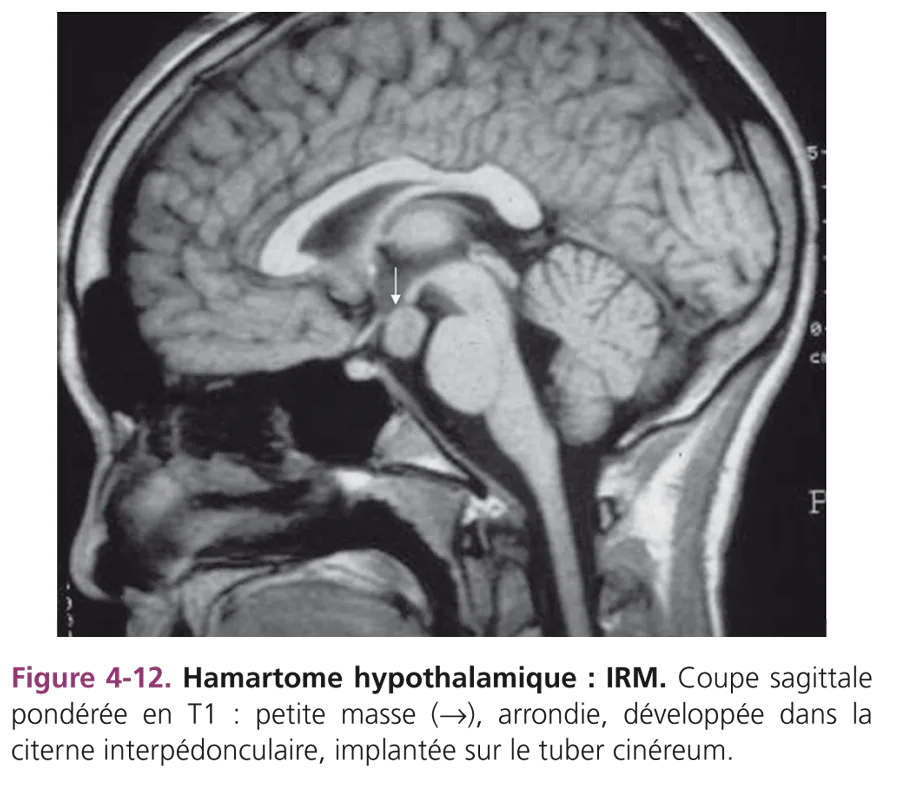
Le bilan biologique étaye le diagnostic en révélant une importante élévation de LH (Luteinizing Hormone) sous stimulation par la GnRH. Le taux d’oestradiol est élevé dans 75 % des cas. L’enquête étiologique systématiquement menée recherche une lésion intracrânienne, idéalement par IRM. Une lésion organique est retrouvée dans moins de 10 % des pubertés centrales chez la fille. Elles sont à craindre lorsque le développement pubertaire survient à un jeune âge. La première cause de puberté précoce centrale par lésion cérébrale est l’hamartome hypothalamique, pseudo-tumeur non évolutive qui se comporte comme un centre ectopique de sécrétion de GnRH. Cette lésion, plus ou moins arrondie, de taille variable, est toujours implantée sur les piliers des tubercules mammillaires, dans la citerne interpédonculaire, en arrière de la tige pituitaire (fig. 4-12). D’autres tumeurs peuvent entraîner une précocité pubertaire : le germinome se développe sur la ligne médiane, dans la région sellaire ou épiphysaire. Le diagnostic est fait par le dosage de β-hCG (hormone chorionique gonadotrope) dans le sang et le LCS (liquide cérébrospinal). C’est cette sécrétion anormale qui provoque le développement pubertaire.
Les gliomes sont possibles, survenant isolément ou souvent dans le cadre d’une neurofibromatose. Les autres causes sont résumées dans l’encadré 4-2.

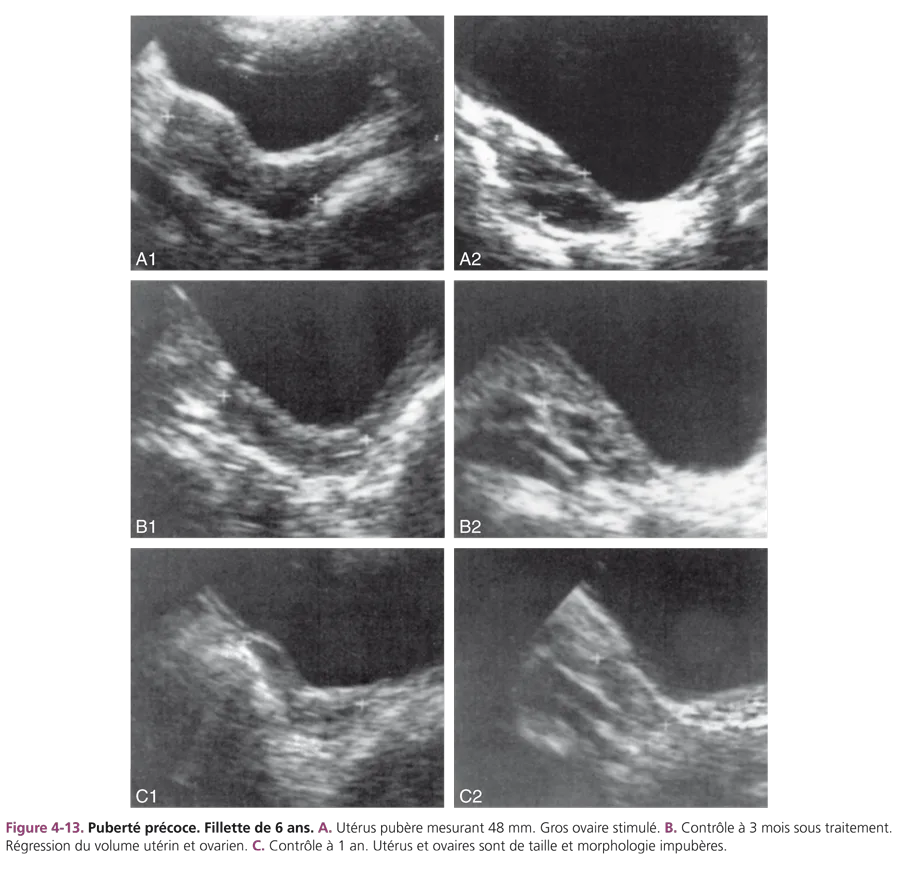
Le traitement de la puberté précoce idiopathique ou liée à un hamartome est médical, freinateur, par administration d’agonistes de la GnRH. Ce traitement, très efficace, entraîne une freination pubertaire prolongée, sans échappement [6]. L’échographie de surveillance, si elle est effectuée, constate la réduction du volume ovarien et l’absence d’activité folliculaire (fig. 4-13). La régression utérine est souvent partielle. À l’arrêt du traitement, le développement pubertaire s’effectue normalement.
Pubertés précoces périphériques Bien plus rares, elles sont d’origine ovarienne ou surrénalienne, liées à une sécrétion autonome. Elles se traduisent par un développement pubertaire dysharmonieux, une accélération de la vitesse de croissance et de la maturation osseuse. En cas de sécrétion autonome ovarienne, la poussée mammaire est franche avec des aréoles souvent pigmentées et parfois des métrorragies. Il n’y a pas de signe androgénique franc. À l’inverse, la sécrétion prématurée d’androgènes d’origine surrénalienne se marque par l’apparition prématurée et isolée d’une pilosité pubienne, d’une acné et d’une hypertrophie clitoridienne.
Syndrome de Mac Cune-Albright [7] Il associe typiquement une précocité pubertaire dissociée d’origine ovarienne, des taches pigmentaires cutanées et une dysplasie fibreuse des os. Le taux d’œstradiol sanguin est souvent très élevé avec un test de stimulation à la GnRH plat, témoin du dysfonctionnement primitif ovarien. L’échographie retrouve un développement évident des organes génitaux internes. Les ovaires bien développés présentent très souvent des kystes uni- ou bilatéraux (fig. 4-14) qui peuvent être ponctionnés s’ils sont douloureux. Ces kystes sont parfois hémorragiques prenant alors une échostructure solide, hétérogène et leur découverte initiale peut simuler alors une tumeur sécrétante de l’ovaire. Un contrôle échographique à distance permet de rectifier le diagnostic en visualisant les modifications du contenu du kyste.
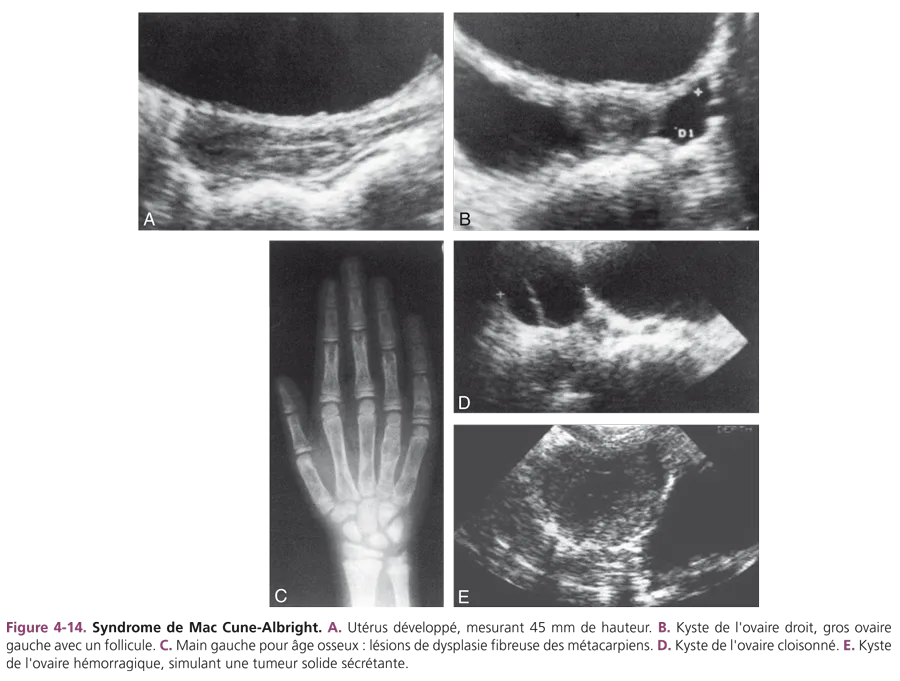
Des radiographies du squelette complet recherchent des lésions de dysplasie fibreuse des os. Le syndrome de Mac Cune-Albright est lié à une pathologie des récepteurs membranaires (anomalie de la protéine G), qui entraîne un syndrome d’autonomisation polyorganique. D’autres endocrinopathies (atteinte thyroïdienne, atteinte surrénalienne et hypophysaire) peuvent s’associer.
Tumeur ovarienne sécrétante Leur diagnostic est échographique, effectué sur la présence d’une masse ovarienne, d’aspect non spécifique, souvent hétérogène et volumineuse, complété par un scanner pour le bilan d’extension. La tumeur de la granulosa est la plus fréquente.
Puberté précoce à forme virilisante Leur origine est surrénalienne et le premier diagnostic à éliminer est un corticosurrénalome, facilement identifié à l’échographie avant 6 ans. Il s’agit d’une tumeur souvent bien limitée, de taille variable et de structure assez homogène. Chez l’enfant plus grand, l’exploration par tomodensitométrie ou IRM est recommandée afin de ne pas méconnaître une petite tumeur. Dans ce contexte, l’autre diagnostic à éliminer biologiquement est une hyperplasie congénitale des surrénales par bloc enzymatique.
Variantes du développement pubertaire ou puberté dissociée [5] Ces situations cliniques sont fréquentes et doivent être distinguées des états pathologiques précédemment décrits : le développement pubertaire ne porte que sur un seul caractère sexuel secondaire. L’avance staturale parfois constatée reste discrète et l’âge osseux n’excède jamais l’âge réel.
Développement mammaire isolé ou prémature thélarche Il survient souvent chez les petites filles de 6 mois à 3 ans. L’examen échographique montre un utérus et des ovaires d’aspect impubère, normal pour l’âge. Les ovaires de petite taille peuvent présenter des follicules (fig. 4-15). Le bilan hormonal est normal.
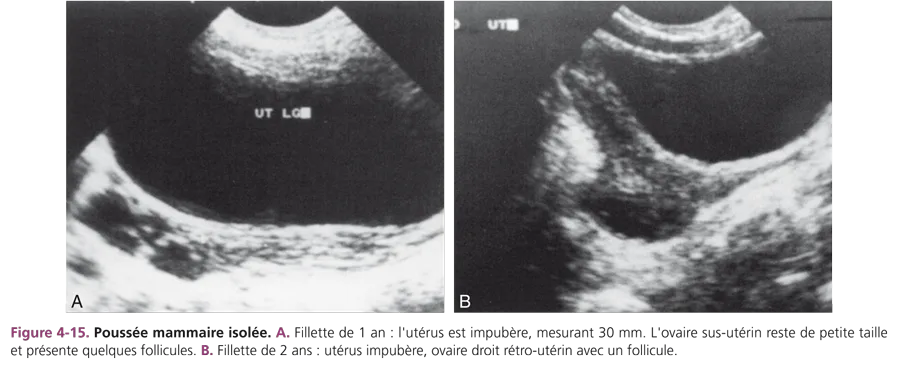
Une surveillance clinique reste indispensable, contrôlant la régression spontanée du symptôme. L’évolution vers une puberté précoce vraie se voit surtout chez la fillette de 4 à 7 ans.
L’hypothèse d’un excès de réceptivité aux oestrogènes des organes cibles est évoquée. Le même mécanisme expliquerait les métrorragies isolées.
Métrorragies isolées ou prémature ménarche
Elles surviennent chez des fillettes sans aucun développement pubertaire. L’échographie est essentielle : elle montre l’absence de développement utéro-ovarien, éliminant une tumeur de l’ovaire ou un syndrome d’autonomisation ovarienne. Elle vérifie l’absence de corps étranger intravaginal [8] et l’absence de rhabdomyosarcome du sinus urogénital. C’est donc un diagnostic d’élimination et la surveillance clinique seule est proposée.
Précocité pileuse ou prémature adrénarche
Elle est assez fréquente chez les filles de 6 à 7 ans impubères. L’examen ultrasonore confirme l’absence de développement utéro-ovarien. Une échographie surrénalienne est de réalisation indispensable afin d’éliminer un corticosurrénalome. Les dosages hormonaux (lignée des androgènes) seront systématiquement réalisés. Le suivi simple des enfants constate une puberté ultérieure normale.
Aménorrhée primaire
Elle se définit par l’absence de règles à 15 ans chez une fille. Deux situations doivent être différenciées en fonction de la présence ou pas d’un retard pubertaire.
Sans retard pubertaire
L’échographie pelvienne tient une place prépondérante en montrant souvent une malformation utérovaginale. L’hématocolpos est de diagnostic aisé devant des douleurs pelviennes : il s’agit d’une masse liquidienne, à contenu souvent échogène ou hétérogène. Cette masse est médiane et plonge sous la symphyse pubienne. L’utérus sus-jacent est normal ou peut présenter une hématométrie. L’obstacle peut être distal par imperforation hyménéale. Plus rarement, il s’agit d’un diaphragme ou d’une aplasie vaginale plus ou moins étendue.
Le syndrome de Mayer-Rokitansky-Küster-Hauser associe une aplasie plus ou moins complète du vagin et un utérus rudimentaire, parfois bicorne. Les trompes sont souvent normales, parfois hypoplasiques. Des malformations rénales sont présentes dans 30 à 40 % des cas : agénésie rénale unilatérale, reins fusionnés, rein pelvien, reflux vésico-urétéral. Des anomalies osseuses vertébrales ou costales sont associées dans 10 % des cas. L’insensibilité complète aux androgènes ou syndrome de féminisation testiculaire est une cause exceptionnelle d’aménorrhée primaire. L’échographie ne trouve aucune structure utéro-ovarienne. Le vagin est court et borgne. Le caryotype est XY. Les testicules peuvent être palpés dans l’aine. Le taux de testostérone est normal pour un sujet XY. Les autres étiologies d’aménorrhée primaire recoupent celles des retards pubertaires dans leur forme incomplète.
Avec retard pubertaire
Il n’existe pas de caractère sexuel secondaire. Les données cliniques et le bilan hormonal permettent de distinguer les hypogonadismes hypogonadotrophiques d’origine centrale des hypogonadismes hypergonadotrophiques d’origine ovarienne dont les étiologies sont résumées dans l’encadré 4-3.

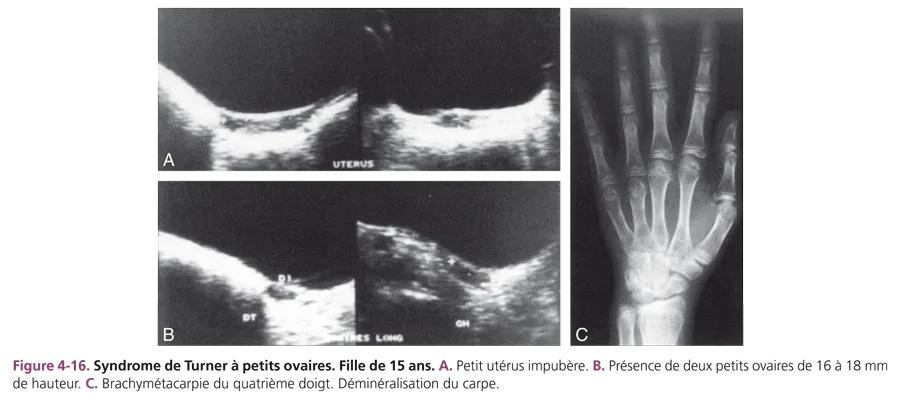
Les hypogonadismes d’origine centrale peuvent se traduire par un impubérisme plus ou moins complet. L’échographie retrouve des organes génitaux internes impubères. L’imagerie recherche une cause centrale et notamment une tumeur de la région sellaire : craniopharyngiome, gliome associé ou non à une neurofibromatose de type I, voire adénome à prolactine. Le syndrome de Kallmann ou dysplasie olfactogénitale est une malformation associant un retard pubertaire et une anosmie ou hyposmie. En IRM, l’antéhypophyse peut être de petite taille et l’hypoplasie (ou aplasie) des bulbes et des sillons olfactifs est objectivée par des coupes fines centrées. L’insuffisance gonadotrope peut s’intégrer dans un panhypopituitarisme congénital. Les hypogonadismes d’origine ovarienne peuvent correspondre à une dysgénésie gonadique dans le cadre d’un syndrome de Turner. Le retard statural y est constant et s’associe à des signes typiques de dysmorphie : cou court avec ptérygium coli, écartement mamelonnaire, cubitus valgus. Les signes osseux classiques sont la brachymétacarpie des IVe et Ve rayons (fig. 4-16C), une trame osseuse grossière et une hypertrophie du condyle interne du genou (signe de Kosowicz). L’examen échographique montre un utérus impubère et des ovaires non individualisables, car ils sont constitués de bandelettes fibreuses, sans follicule. Rarement, les ovaires sont visibles, de petite taille (fig. 4-16A et B). Une échographie rénale doit être couplée à l’étude pelvienne à la recherche de malformations rénales souvent associées (rein en fer à cheval, bifidité, hypotonie pyélo-urétérale, hypoplasie rénale unilatérale). Soixante pour cent des syndromes de Turner présentent une monosomie 45X0 et il existe des mosaïques.
Anomalies de la différenciation sexuelle (Disorders of Sex Development [DSD])
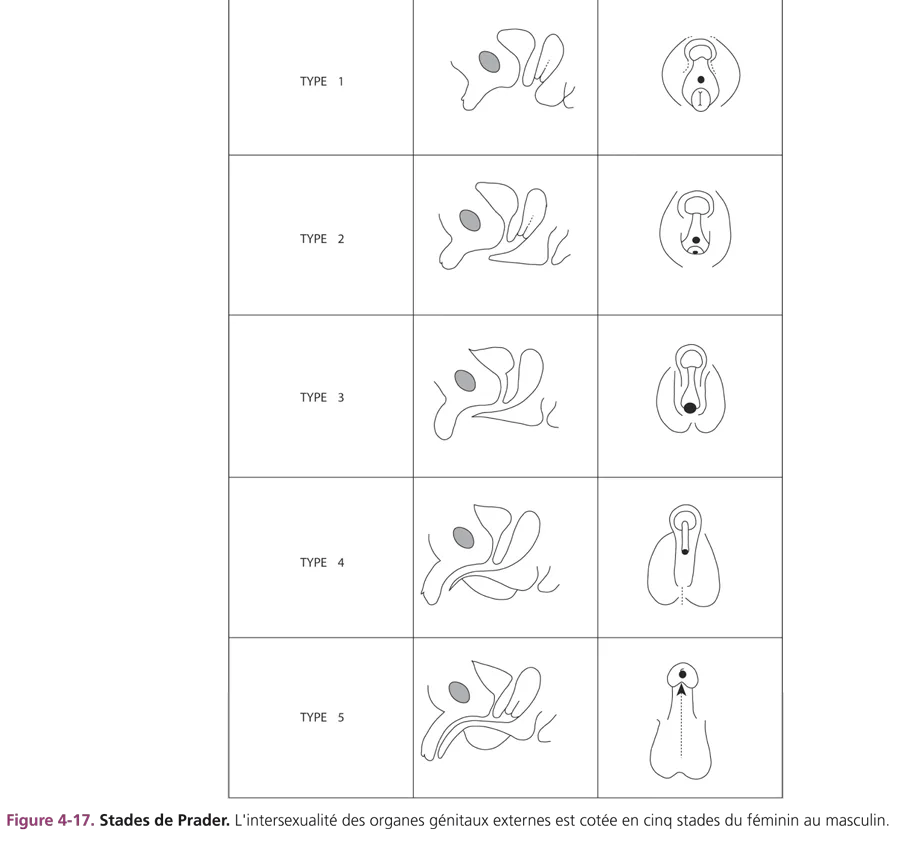
Leur diagnostic se pose à la période néonatale devant un enfant présentant une malformation des organes génitaux externes. Cette situation clinique impose un diagnostic étiologique rapide et une évaluation des possibilités de chirurgie réparatrice afin d’envisager le choix du sexe. L’examen clinique constate un développement plus ou moins marqué du bourgeon génital, la présence d’un ou de deux orifices périnéaux, la topographie plus ou moins postérieure de l’hypospade et la soudure plus ou moins complète des bourrelets labioscrotaux dans lesquels on peut palper des gonades. Le degré de différenciation des organes génitaux externes est coté selon les stades de Prader (fig. 4-17).
Le bilan biologique initial comporte un ionogramme sanguin et des dosages hormonaux, notamment de la 17-hydroxy-progestérone. Le caryotype est prélevé. La chromatine sexuelle est rapidement obtenue. L’échographie pelvienne peut montrer la présence d’un utérus qui oriente d’emblée vers le diagnostic de DSD XX. La génitographie est différée par rapport au bilan initial. Elle évalue le degré de la malformation interne : longueur de l’urètre antérieur, présence d’une cavité postérieure de taille variable, implantée plus ou moins haut sur l’urètre postérieur. L’incidence de profil strict est indispensable. Un cliché de face est toujours effectué, montrant si cette cavité est unique ou double, médiane ou latérodéviée [9].
L’étude échographique des gonades est souvent difficile et non contributive. Il existe deux grands cadres nosologiques. Les DSD sont définis comme un état pathologique dans lequel le sujet est porteur de gonades différenciées dans un sexe et dont les organes génitaux externes sont plus ou moins développés dans le sexe opposé : virilisation d’une fille ou défaut de masculinisation d’un garçon. Les autres états intersexuels correspondent à des anomalies de la différenciation gonadique d’origine chromosomique ou génique. Dans cette indication, l’échographie par voie suspubienne peut être complétée par voie périnéale (cf. infra DSD XY).
Anomalies de la différenciation sexuelle XX (DSD XX)
Elles représentent environ 40 % des états intersexuels. Ce sont des filles, au caryotype 46XX, présentant une virilisation plus ou moins marquée du sinus urogénital. Les ovaires sont normaux et en place. L’utérus est présent. Le vagin présente un orifice dont l’implantation est variable (fig. 4-18 et 4-19) [10,11].
L’étiologie est l’hyperplasie congénitale des surrénales par bloc enzymatique. Le bloc en 21-hydroxylase est le plus fréquent et son marqueur biologique est une importante élévation de la 17-hydroxy-progestérone. Les androgènes surrénaliens accumulés en amont du bloc virilisent le foetus femelle. En aval, le déficit en gluco- et minéralocorticoïdes est cliniquement évident et nécessite un traitement substitutif. Exceptionnellement, la virilisation peut être liée à la prise inappropriée d’androgènes par la mère pendant la grossesse.
Anomalies de la différenciation sexuelle XY (DSD XY)
Elles représentent environ 45 % des états intersexuels. Ce sont des garçons, au caryotype normal 46XY, présentant des testicules normaux souvent palpés dans les bourrelets labioscrotaux et dont la masculinisation du sinus urogénital est insuffisante. Le taux sanguin de testostérone est normal. L’échographie montre l’absence de structure utérine puisqu’elle a régressé sous l’effet de l’hormone antimüllérienne normale. La génitographie révèle souvent la présence d’une cavité postérieure qui témoigne du défaut de masculinisation du sinus urogénital. L’insensibilité aux androgènes est l’étiologie principale, recherchée par l’étude des fibroblastes de la peau périnéale biopsiée. L’insensibilité est liée à un trouble de réceptivité aux androgènes par anomalie quantitative ou qualitative des récepteurs dans les tissus cibles, souvent difficile à mettre en évidence. Sa forme complète réalise le syndrome de féminisation testiculaire. Dans les formes incomplètes, le trouble de la différenciation sexuelle est plus ou moins sévère, posant de difficiles problèmes de choix du sexe (fig. 4-20) car l’insensibilité persistera à la période pubertaire.
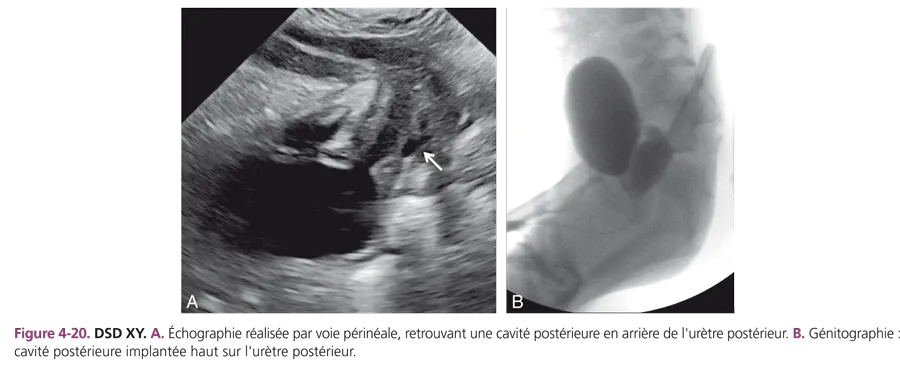
Beaucoup plus rarement, il s’agit d’un déficit en 5-alpharéductase, enzyme intracellulaire qui convertit la testostérone en dihydrotestostérone (DHT) active. Enfin, il peut s’agir d’un exceptionnel déficit enzymatique dans la chaîne de synthèse de la testostérone.
Dysgénésie gonadique mixte ou asymétrique
Elle représente environ 12 % des états intersexuels. Le caryotype typique est une mosaïque 45XO/46XY. Le plus souvent, il existe un testicule dysgénétique palpé en situation inguinale ou scrotale et du côté opposé, siège de la régression gonadique, un utérus normal ou hypoplasique latérodévié dans le pelvis à l’échographie et sur la génitographie. Le risque de dégénérescence gonadique impose la castration avec mise en place d’un traitement substitutif à la puberté.
DSD ovo-testiculaire
Il représente 3 % des états intersexuels et se définit par la présence chez un même sujet de tissu ovarien et testiculaire. Le caryotype est 46XX dans 50 % des cas, 46XY dans 25 % des cas ou une mosaïque. Le risque de dégénérescence est faible.
Masses pelviennes
Les circonstances de découverte d’une masse pelvienne sont variées [12–14] : ■ palpation d’une masse chez un enfant asymptomatique ; ■ signes cliniques orientant vers une tumeur : puberté précoce, aménorrhée primaire et douleurs pelviennes, signes d’hyperandrogénie (acné, hirsutisme) ; ■ découverte fortuite lors d’une échographie ; ■ mise en évidence lors d’une échographie anténatale.
L’approche diagnostique repose surtout sur le couple échographie abdominopelvienne-IRM et les marqueurs tumoraux (α-foetoprotéine, β-hCG…). Le cliché d’abdomen de face peut mettre en évidence des calcifications intratumorales, un contingent graisseux et/ou des anomalies osseuses du bassin et du rachis. L’échographie effectuée vessie pleine affirme la présence d’une masse, en précise sa topographie, sa taille et ses contours, l’échogénicité de son contenu et si possible les rapports aux organes adjacents. Ces données confrontées à celles de l’âge et du sexe de l’enfant sont souvent suffisantes au diagnostic. L’IRM est indispensable à la recherche d’un prolongement intrarachidien d’une tumeur postérieure et au bilan d’extension d’une masse du plancher pelvien. L’injection de produit de contraste paramagnétique est systématique. Si la lésion est maligne, l’examen tomodensitométrique reste fondamental dans le bilan d’extension locorégionale (ganglions, vaisseaux, parties molles, os).
Masses pelviennes de la période néonatale
Elles sont listées selon leur échostructure dans le tableau 4-2. Elles sont très souvent dépistées actuellement par l’échographie anténatale.
Si la masse est liquidienne, il faut éliminer d’emblée une anomalie urinaire : grosse vessie (par immaturité vésicale transitoire ou réflexe par la prise de myorelaxants par la mère ou dans le cadre d’un syndrome mégavessie-microcôlon), diverticule vésical volumineux, méga-uretère congénital, rein multikystique étendu au pelvis. L’énumération de ces diagnostics différentiels souligne la nécessité d’examiner l’appareil urinaire lorsque l’échographie pelvienne décèle une masse ou une malformation. Le repérage du rectum, en échographie au moins, est également un point important.
Kyste de l’ovaire
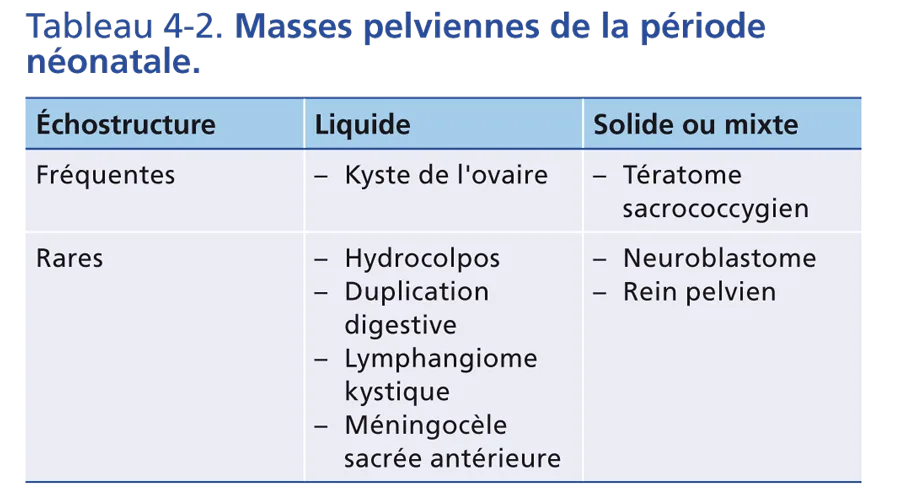
Chez un nouveau-né de sexe féminin, une masse liquidienne est un kyste de l’ovaire jusqu’à preuve du contraire. L’échographie est pratiquée pour confirmer un diagnostic anténatal ou pour étudier une masse abdominopelvienne palpée, ces kystes pouvant être très volumineux. Cliniquement, les kystes ovariens sont souvent latéralisés et mobiles, de topographie abdominopelvienne ou parfois abdominale, controlatérale à la situation d’origine. Il s’agit de kystes folliculaires simples, liquidiens, transsonores, à parois fines, de taille variable. Si le kyste est supérieur à 4 cm, le risque de torsion incite à pratiquer une ponction en période anténatale ou à la naissance (fig. 4-21).
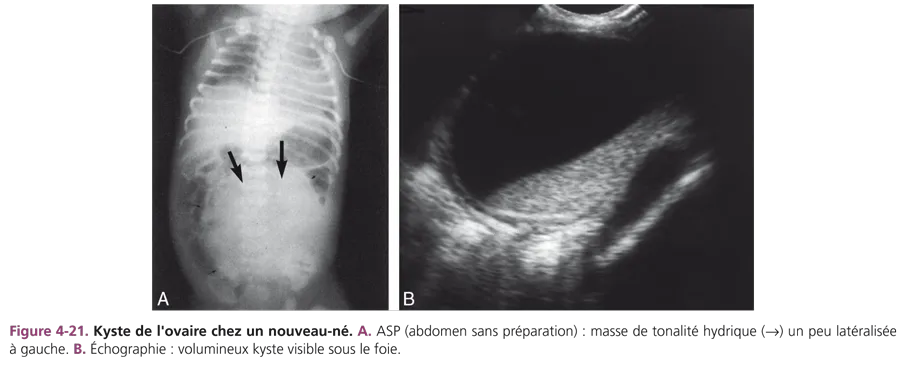
En dehors de la torsion, l’évolution est spontanément favorable avec régression du kyste en moins de 6 mois. La récidive possible du kyste pendant environ 1 mois sous l’effet de la stimulation hormonale maternelle justifie une surveillance échographique. Les kystes ovariens peuvent présenter d’emblée un aspect trompeur d’échostructure hétérogène avec des échos de répartition irrégulière ou déclives avec niveau liquide ou d’échostructure homogène, échogène donnant un aspect de masse solide (fig. 4-22A). Ces aspects sont liés à une hémorragie intrakystique spontanée, traumatique ou secondaire à une torsion du fait de l’étirement de la trompe adjacente. Le diagnostic de torsion est difficile au Doppler car l’altération du flux n’est pas toujours appréciée de façon fiable (fig. 4-22B). Ces kystes hémorragiques sont en général opérés, bien que la récupération de l’ovaire soit aléatoire.
Hydrocolpos ou hydrométrocolpos
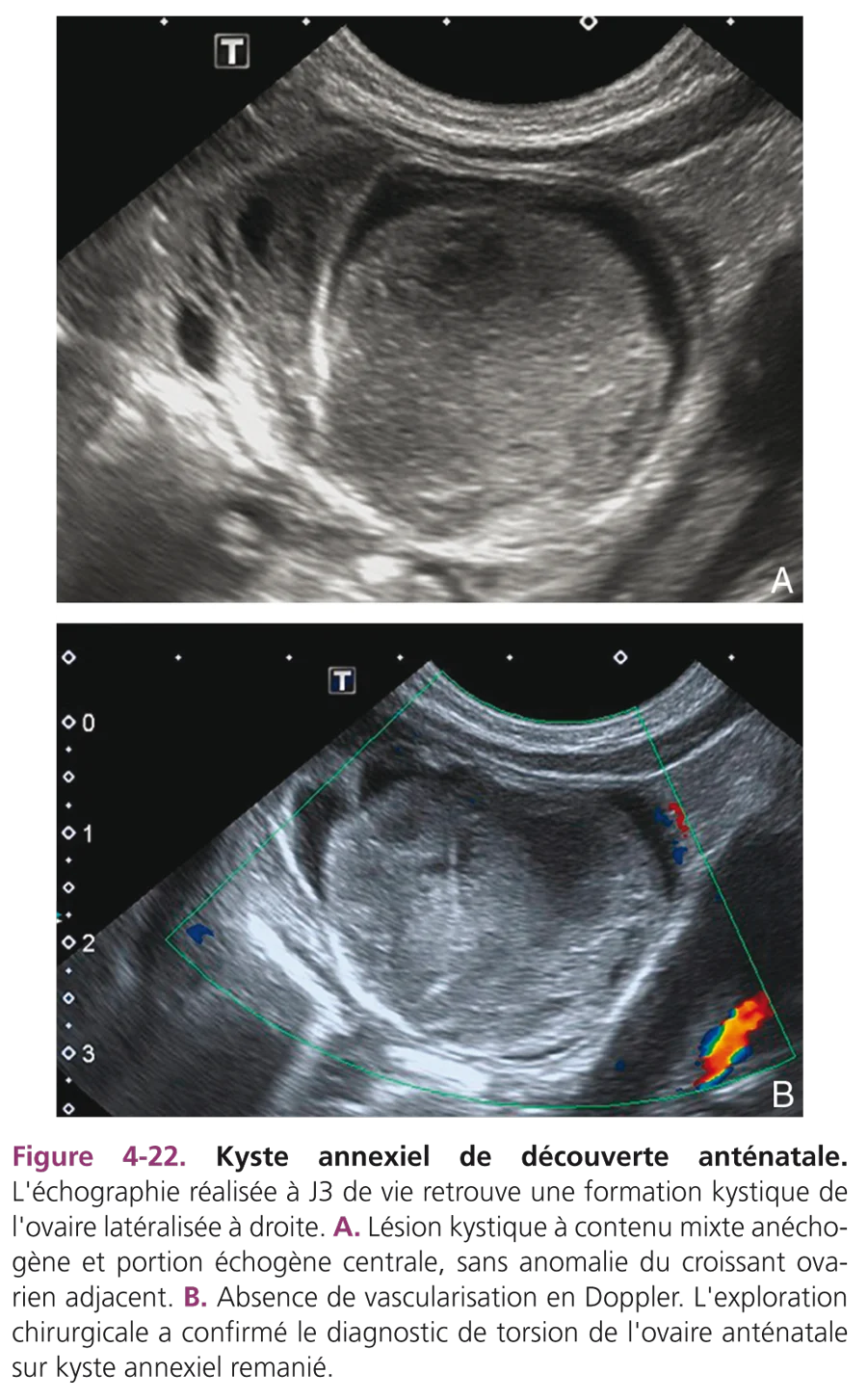
Il est secondaire à une malformation vaginale plus ou moins complexe. Il est souvent découvert à la naissance dans un tableau clinique de polymalformations : imperforation anale, malformation urogénitale, cardiopathie, polydactylie. Cliniquement et échographiquement, l’hydrocolpos diffère du kyste de l’ovaire car il est médian, fixe et plonge sous la symphyse pubienne (fig. 4-23). La rétention est rarement liquidienne pure, elle contient souvent des mucosités formant un sédiment déclive ou homogène. La cavité utérine sus-jacente peut être dilatée ou non. Dans le cadre d’une duplicité, la malformation associe classiquement un hémihydrocolpos et une agénésie rénale unilatérale.
Tératome sacrococcygien
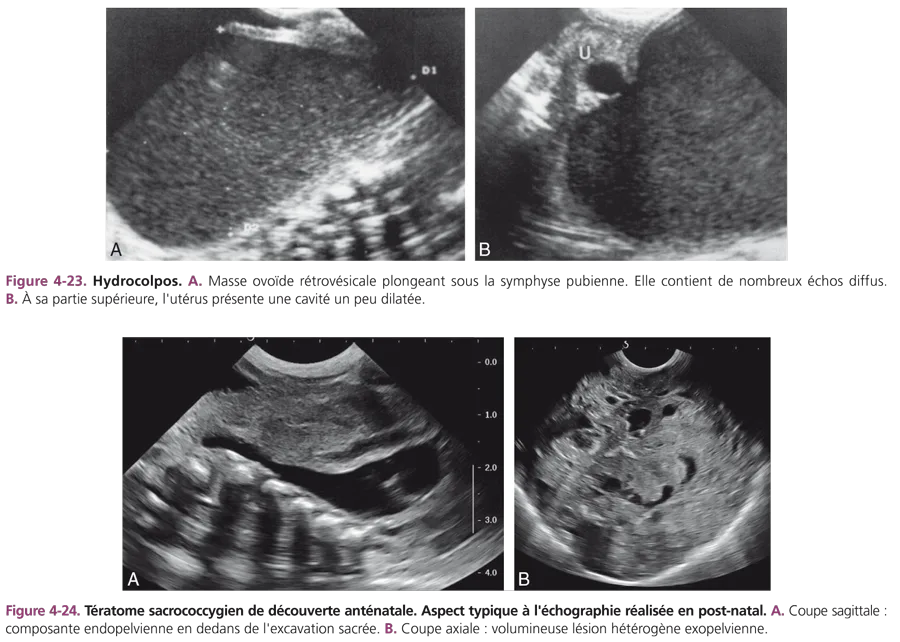
Peu fréquente, cette tumeur touche quatre fois plus souvent les filles que les garçons. Son diagnostic est actuellement anténatal. À la naissance, il s’agit d’une masse sacrococcygienne, extériorisée à la fesse et/ou au périnée. Dix pour cent de ces tumeurs ont un développement purement endopelvien, présacré, rétrorectal. Bien que 95 % des tératomes sacrococcygiens soient bénins, le dosage de l’alpha-foetoprotéine et des gonadotrophines sériques doit être pratiqué, avant toute intervention chirurgicale d’exérèse. Le cliché d’ASP est caractéristique lorsqu’il montre des malformations vertébrales sacrées et des calcifications intratumorales. L’échostructure de la masse intra- et extrapelvienne est hétérogène, contenant des éléments liquidiens et solides (fig. 4-24). L’examen tomodensitométrique montre les contingents graisseux et les petites calcifications. L’IRM précise l’extension périnéale et intrarachidienne par les trous sacrés.
À la naissance, la quasi-totalité des tératomes sacrococcygiens étant bénins, le traitement est uniquement chirurgical. La surveillance repose sur les marqueurs et l’échographie. Le diagnostic différentiel se pose avec le neuroblastome pelvien qui est plus rare. C’est une tumeur solide, sans contingent graisseux. Le dosage des catécholamines permet d’en affirmer le diagnostic.
Masses pelviennes en dehors de la période néonatale
Elles sont présentées selon leur échostructure dans le tableau 4-3, trois pièges diagnostiques sont à écarter : un volumineux fécalome, un rein pelvien et une grossesse chez la jeune fille pubère.
Kystes ovariens fonctionnels
Fréquents à la période pubertaire, il s’agit de kystes folliculaires ou lutéiniques dont le diamètre est supérieur à 3 cm. Même s’ils sont volumineux, leur caractère spontanément résolutif autorise la surveillance échographique après un ou deux cycles. Ce n’est qu’en cas de kyste volumineux et persistant qu’un traitement est proposé, soit médical (hormonal), soit par ponction ou coelioscopie. Ces kystes peuvent être d’aspect solide en cas d’hémorragie (kyste du corps jaune hémorragique) et l’évolution se fait vers la lyse progressive du caillot. Un aspect particulier est la présence de « vésicules filles » au sein du kyste.
Torsion d’annexe
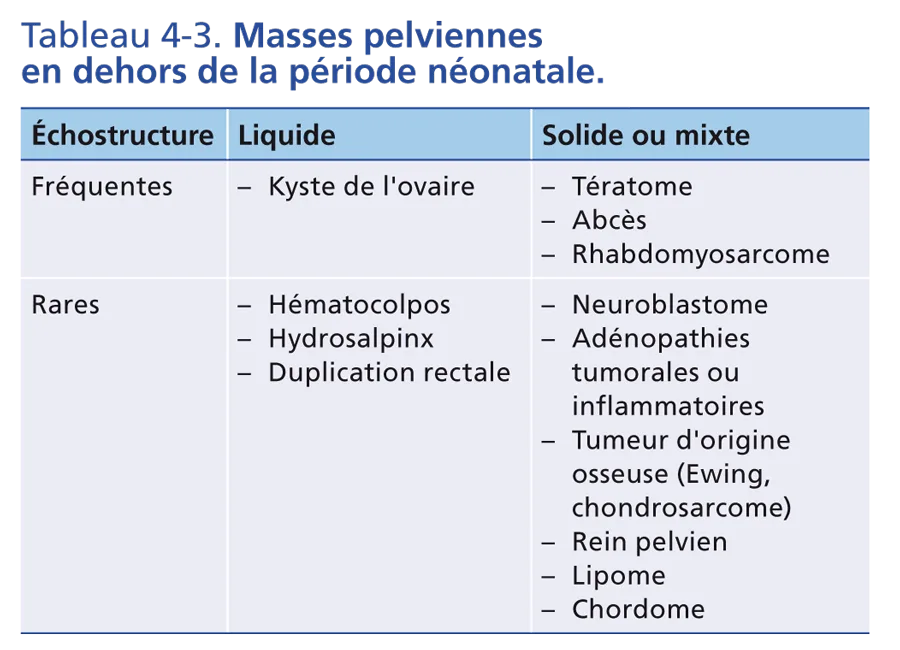
Il s’agit d’une rotation de l’ovaire sur son pédicule vasculaire, dont l’aspect morphologique est variable depuis l’oedème ovarien jusqu’à la nécrose. C’est une urgence chirurgicale. La torsion d’annexe peut être favorisée par un kyste ou une tumeur ovarienne. La présentation clinique classique associe une douleur pelvienne aiguë avec des nausées et/ou des vomissements. Du fait de l’hyperthermie modérée possible et d’une hyperleucocytose, à droite, la torsion d’annexe peut réaliser un tableau pseudoappendiculaire. Les formes subaiguës peuvent faire croire à tort à une tumeur. En échographie, la torsion est marquée par une augmentation de volume de l’ovaire, un épanchement dans le cul-de-sac de Douglas et un élargissement de la trompe (fig. 4-25). La présence de petits kystes centimétriques, localisés en périphérie de la masse, est un critère diagnostique majeur, mais l’aspect est souvent non spécifique, hétérogène avec contingent solide et kystique.
La présence d’un signal Doppler artériel au sein de la masse n’exclut pas le diagnostic. L’IRM a un intérêt en cas de difficulté diagnostique et de formes subaiguës, en montrant les formations kystiques périphériques et l’absence de rehaussement après injection de produit de contraste.
Hématocolpos
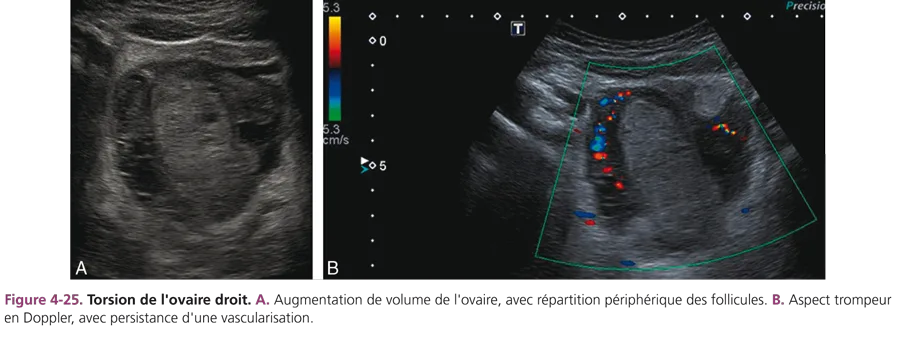
Révélé par une aménorrhée primaire et des douleurs pelviennes, son aspect échographique de rétention vaginale plus ou moins liquidienne est caractéristique (fig. 4-26). L’imperforation hyménéale est classique. Plus rarement, il s’agit d’un diaphragme ou d’une atrésie, notamment dans la forme unilatérale sur duplicité (association avec une agénésie rénale homolatérale).
Tumeurs ovariennes
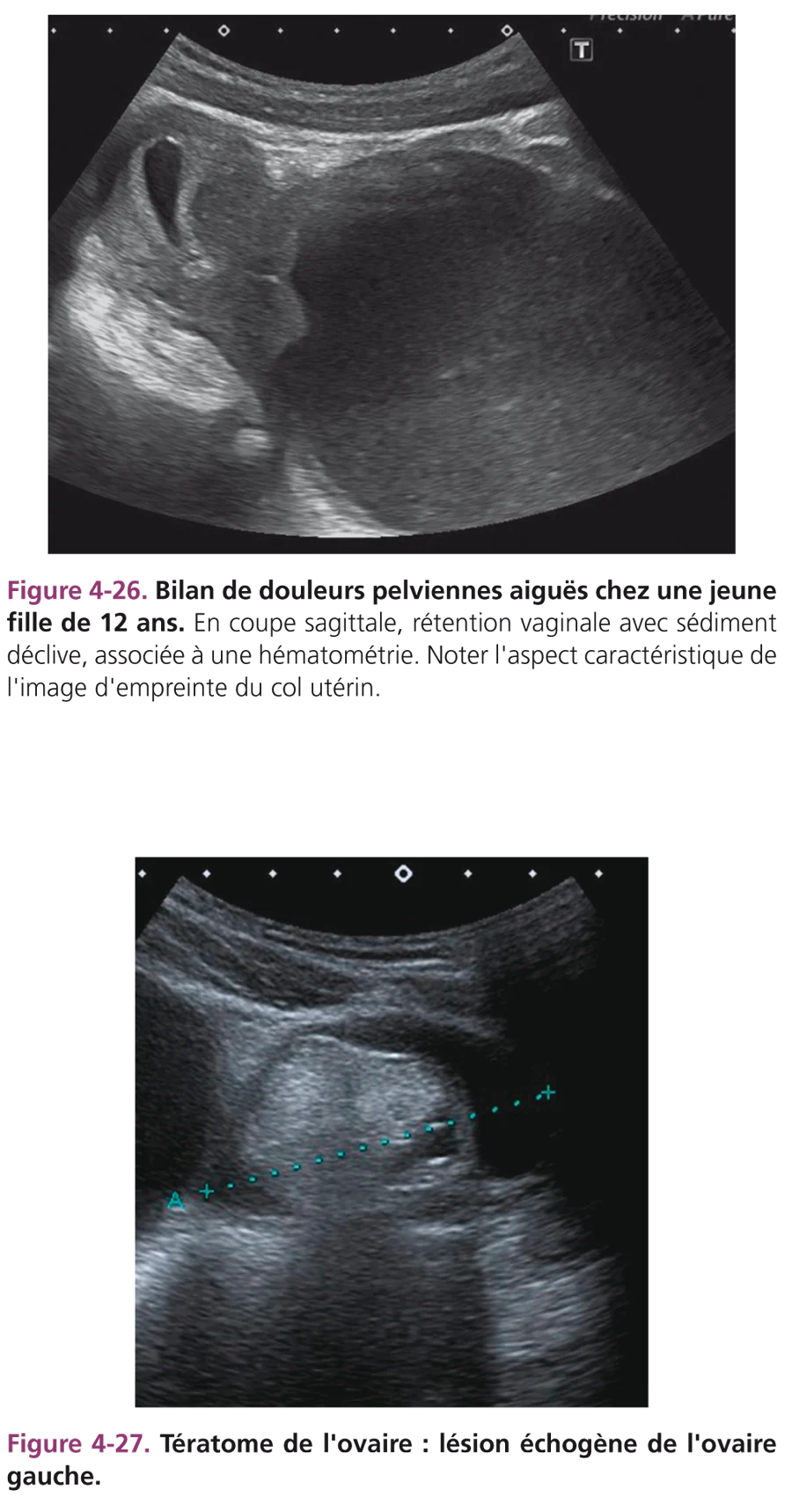
Elles sont rares chez l’enfant et le plus souvent bénignes, représentées essentiellement par des tumeurs germinales. Le tératome mature représente 90 % des tumeurs bénignes. Le tératome mature ou kyste dermoïde est la principale tumeur de l’ovaire chez l’enfant. Il survient principalement entre 6 et 11 ans bien qu’il puisse être observé à tout âge. Environ 10 % de ces tumeurs sont bilatérales. En échographie, la lésion est hétérogène et comporte une partie kystique avec un ou plusieurs nodules muraux échogènes, graisseux et/ou calciques (fig. 4-27). La présence de structure dentaire ou osseuse est très caractéristique. La masse peut contenir un liquide épais, sébacé, échogène. Le scanner ou l’IRM permet d’identifier le contenu calcique et graisseux et de vérifier l’ensemble de la cavité abdominopelvienne (foie, adénopathies, épanchement intrapéritonéal). Le dosage des marqueurs tumoraux est systématiquement effectué (alpha-foetoprotéine et β-hCG). Les tumeurs germinales malignes sont rares et représentent environ 85 % des tumeurs malignes de l’ovaire chez l’enfant. Elles se rencontrent surtout entre 10 et 15 ans. Il s’agit souvent de tumeurs germinales malignes mixtes associant différentes composantes. Les contingents sécrétants sont les tumeurs du sac vitellin (ou tumeurs du sinus endodermique) sécrétant de l’alpha-foetoprotéine et les choriocarcinomes produisant de la β-hCG.
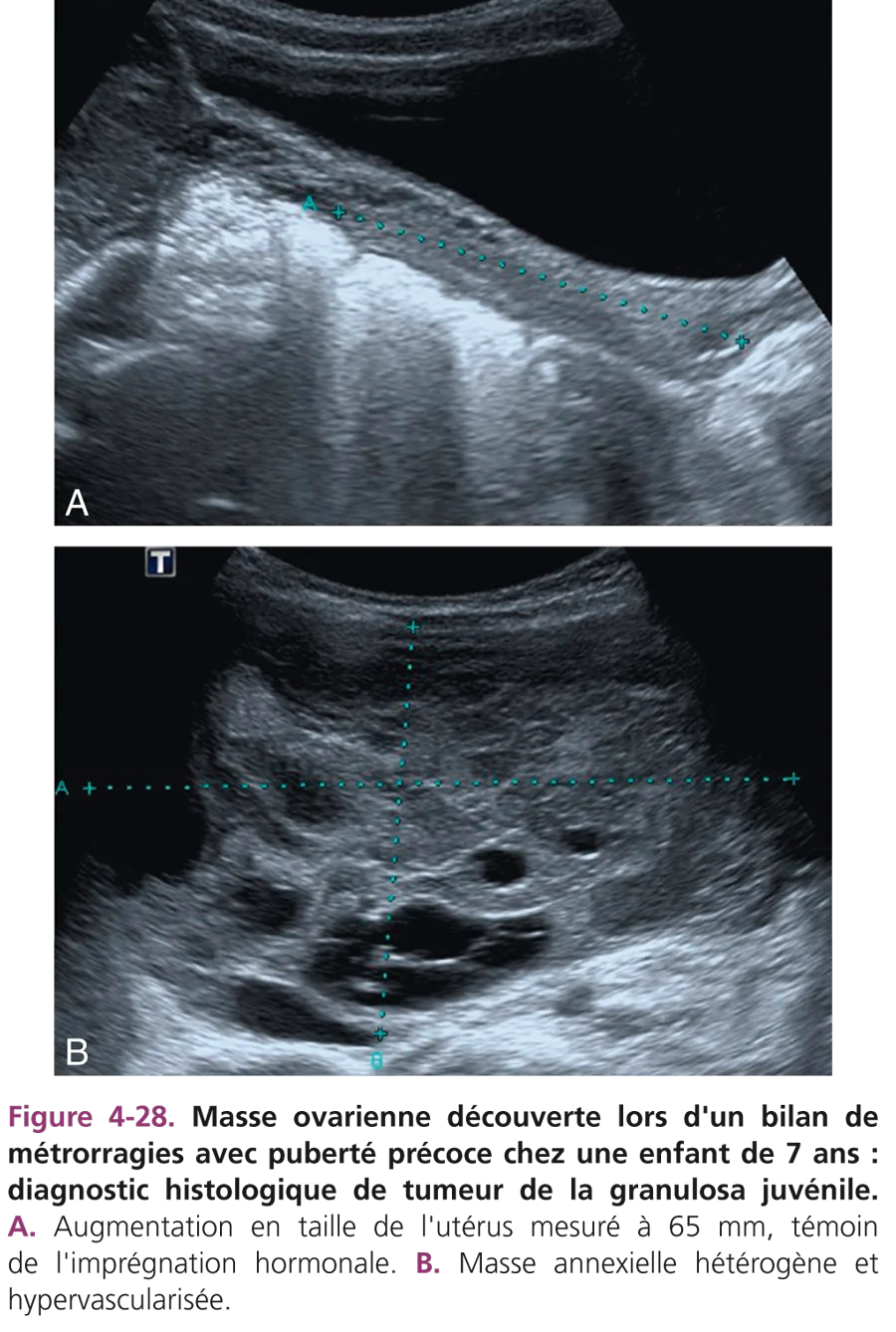
Le dysgerminome est la forme histologique la plus fréquente des tumeurs malignes de l’ovaire à l’âge pédiatrique. Il est, le plus souvent, non sécrétant. Les autres tumeurs ovariennes sont très rares : tumeurs épithéliales (cystadénomes séreux et mucineux), tumeurs de la granulosa avec hyperoestrogénie (fig. 4-28), etc. [14]. Des localisations métastatiques ovariennes peuvent se voir lors des hémopathies malignes.
Abcès pelviens
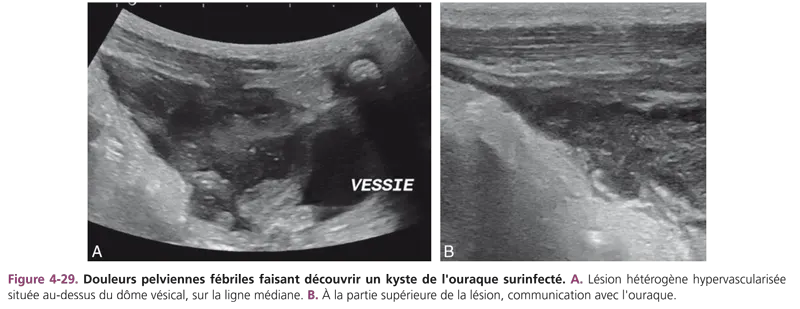
Le plus souvent d’origine appendiculaire, ils peuvent compliquer une infection d’un diverticule de Meckel ou une maladie de Crohn. Ces collections, de topographie intervésicorectale, sont hypoéchogènes, hétérogènes, à paroi épaisse et irrégulière. Il peut s’y associer un épanchement liquidien du cul-de-sac de Douglas. Les données cliniques et biologiques (NFS [numération-formule sanguine], CRP [C-réactive protéine]) aident à porter le diagnostic. En cas de localisation sus-vésicale et médiane de la lésion, il faudra évoquer un kyste de l’ouraque surinfecté (fig. 4-29).
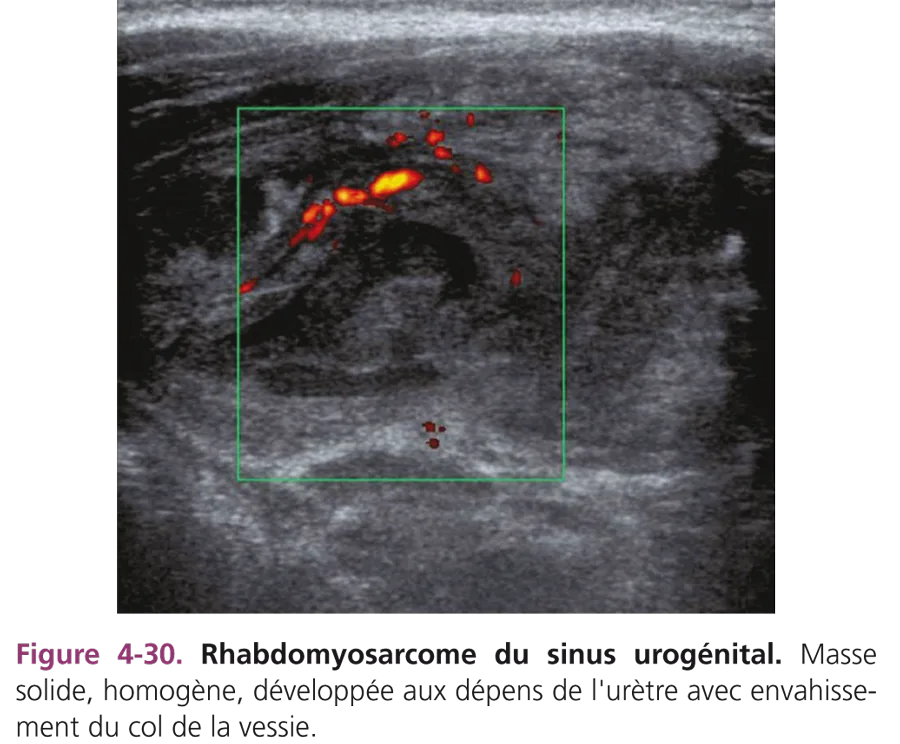
Rhabdomyosarcomes pelviens
Ce sont les tumeurs mésenchymateuses malignes les plus fréquentes chez l’enfant. Les formes pelviennes sont souvent observées dans la petite enfance. Les rhabdomyosarcomes du sinus urogénital se développent aux dépens du trigone vésical, de la prostate, du vagin, de l’utérus et des paramètres. Les signes urinaires d’appel sont fréquents : dysurie, rétention d’urine, hématurie. Parfois, le diagnostic est évoqué devant une masse polypoïde à la vulve (forme botryoïde) chez une fillette ou une grosse bourse chez un petit garçon. L’échographie montre une masse intervésicorectale basse, échogène, lobulée, en apprécie le volume et l’extension à la paroi vésicale (fig. 4-30) et précise le degré d’obstruction du haut appareil urinaire.
Les rhabdomyosarcomes de la paroi pelvienne et du périnée sont souvent très évolués au moment du diagnostic. Le bilan d’extension est en IRM.
Adénopathies tumorales pelviennes
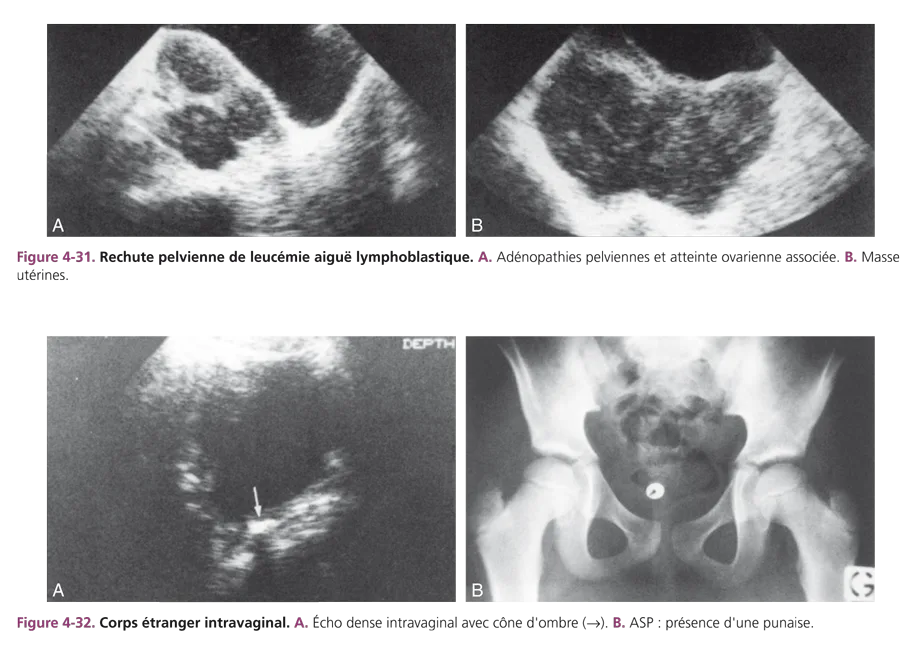
Elles se présentent comme des masses rondes, homogènes, très hypoéchogènes. Elles sont à rechercher lors des rechutes de leucose aiguë. Cette rechute peut intéresser également les ovaires et l’utérus (fig. 4-31). Cette atteinte est méconnue chez la fille alors que la classique rechute testiculaire des garçons est cliniquement évidente.
Au cours des lymphomes non hodgkiniens, les adénopathies iliaques s’associent à une masse iléocæcale et fréquemment à une ascite dont la ponction fait le diagnostic.
Pertes vaginales anormales
Les vulvites sont fréquentes chez la fillette, souvent récidivantes. Les vulvovaginites sont plus rares avec des leucorrhées plus ou moins abondantes, parfois striées de sang. Dans les deux cas, l’infection est liée à des erreurs d’hygiène avec des germes variés non spécifiques. Un corps étranger est à l’origine de 5 % des vulvovaginites, surtout entre 4 et 7 ans. Un cliché d’abdomen sans préparation peut rechercher un corps étranger radio-opaque. L’échographie peut montrer un écho dense intravaginal (fig. 4-32) mais l’examen clinique reste indispensable pour le diagnostic et l’extraction de l’éventuel corps étranger.
L’existence de saignements vaginaux doit faire pratiquer d’emblée une échographie afin de rechercher :
■ un corps étranger intravaginal ; ■ un rhabdomyosarcome du sinus urogénital ; ■ une puberté précoce.
© 2017, Elsevier Masson SAS. Tous droits réservés
Les Auteurs de ce chapitre
S. Chapelière : Chef de clinique assistant, service de radiopédiatrie, CHU du Kremlin-Bicêtre. A. Mathiot : Praticien attaché, service de radiopédiatrie, CHU du Kremlin-Bicêtre. S. Franchi-Abella : Praticien hospitalier, service de radiopédiatrie, CHU du Kremlin-Bicêtre. C. Adamsbaum : Professeur des universités, praticien hospitalier, chef du service de radiopédiatrie, CHU du Kremlin-Bicêtre.
Références
[1] Griffin IJ, Cole TJ, Duncan KA, et al. Pelvic ultrasound measurements in normal girls. Acta Paediatr 1995 ; 84 : 536–43. [2] Haber HP, Mayer EI. Ultrasound evaluation of uterine and ovarian size from birth to puberty. Pediatr Radiol 1994 ; 24 : 11–3. [3] André Ch, Bouvattier-Morel C, Ferey S, et al, editors. Développement pubertaire chez la fille. Aspects normaux et pathologiques. Adamsbaum C. Imagerie Pédiatrique et Foetale. Paris : Médecine- Sciences – Flammarion ; 2007. p. 739–48. [4] Carel JC, Leger J. Clinical practice : precocious puberty. N Engl J Med 2008 ; 358 : 2366–77. [5] Holm K, Laursen EM, Brocks V, et al. Pubertal maturation of internal genitalia : an ultrasound evalulation of 166 healthy girls. Ultrasound Obstet Gynecol 1995 ; 6 : 175–81. [6] Garel L, Dubois J, Grignon A, et al. US of the pediatric female pelvis : a clinical perspective. Radiographics 2001 ; 21 : 1393–407. [7] de Vries L, Phillip M. Role of pelvic ultrasound in girls with precocious puberty. Horm Res Paediatr 2011 ; 75 : 148–52. [8] Caspi B, Zalel Y, Katz Z, et al. The role of sonography in the detection of vaginal foreign bodies in young girls : the bladder indentation sign. Pediatr Radiol 1995 ; 25(S1) : S60–1. [9] André Ch, Beaudoin S, Millischer-Bellaïche AE, et al. Malformations utérovaginales. In : Adamsbaum C, editor. Imagerie Pédiatrique et Foetale. Paris : Médecine-Sciences – Flammarion ; 2007. p. 749–63. [10] Garel L. Abnormal sex differenciation : who, how and when to image. Pediatr Radiol 2008 ; S3 : S508–11. [11] Bouvattier C, David M, Gay CL, et al. Conduite à tenir devant une anomalie des organes génitaux externes découverte à la naissance. Arch Pediatr 2009 ; 16 : 585–7. [12] Peroux E, Franchi-Abella S, Sainte-Croix D, et al. Ovarian tumors in children and adolescents : a series of 41 cases. Diagn Interv Imaging 2015 ; 96 : 273–82. [13] Martelli H, Patte C. Gonadal tumors in children. Arch Ped 2003 ; 10 : 246–50. [14] André C, Brisse H, Adamsbaum C, et al. Masses pelviennes. In : Adamsbaum C, editor. Imagerie Pédiatrique et Foetale. Paris : Médecine-Sciences – Flammarion ; 2007. p. 764–73.
Vous venez de lire un extrait de la sixième édition de l’ouvrage Échographie et imagerie pelvienne en pratique gynécologiqueopens in new tab/window de Yves Ardaens, Jean-Marc Levaillant, Philippe Coquel, Thierry Haag, Bernard Guérin du Masgenêt, Julien Bigot